Mastering Electrophoresis: Techniques, Equipment, and Applications in DNA, RNA, and Protein Analysis

Written By: Craig Bradley
Sep 11, 2025

GEMINI (2025)
In the fields of molecular biology, genetics, and biochemistry, the ability to separate and analyze biological macromolecules is a fundamental requirement for accurate research, diagnostics, and quality control. At the core of this capability lies electrophoresis, a powerful and versatile technique that has become indispensable in the modern laboratory workflow. The principle of electrophoresis involves the migration of charged molecules in a liquid or semisolid medium under the influence of an electric field. By leveraging differences in molecular properties such as size, charge, and shape, this method provides a high-resolution means to purify and characterize nucleic acids and proteins.
This article provides an in-depth examination of the core principles, essential equipment, and a wide array of applications of electrophoresis. A firm grasp of these concepts is crucial for laboratory professionals seeking to ensure data integrity, optimize experimental design, and accelerate scientific discovery. From the foundational theory to the practical execution of techniques for DNA, RNA, and protein analysis, a comprehensive understanding of electrophoresis enables more efficient and reliable laboratory operations.
Core Principles of Electrophoresis: The Science of Molecular Separation
The effectiveness of electrophoresis is rooted in a fundamental physical principle: charged particles will migrate toward an electrode of the opposite polarity when subjected to an electric field. The rate of migration of a molecule, or its electrophoretic mobility, is not uniform. It is governed by a complex interplay of several factors, primarily the molecule's net charge, its size and shape, and the properties of the separation matrix.
The relationship between these factors is described by the equation for electrophoretic mobility
where:
v is the velocity of the molecule.E is the electric field strength.q is the net charge of the molecule.f is the frictional coefficient, a measure of the molecule's resistance to movement through the medium.
A molecule's net charge is a critical determinant of its direction and speed of movement. For nucleic acids, such as DNA and RNA, the phosphate backbone imparts a strong, uniform negative charge, ensuring that all fragments migrate towards the positive anode. This simplifies the process, as the primary separation variable becomes the size of the molecule. In contrast, proteins possess varying charges determined by their amino acid composition and the pH of the buffer, necessitating more complex techniques to achieve uniform migration.
The separation matrix, typically a porous gel, acts as a molecular sieve. The gel's pore size significantly influences the frictional coefficient. Smaller molecules can navigate the gel's pores more easily, experiencing less friction and thus migrating faster. Larger molecules, however, are impeded, resulting in slower movement. This size-based separation is the cornerstone of most electrophoretic applications. The choice of gel material and concentration is paramount to achieving optimal resolution for the target molecules. For instance, low-concentration gels are suitable for separating large molecules, while high-concentration gels are required for resolving smaller molecules.
Mastering Agarose Gel Electrophoresis for DNA and RNA Analysis
Agarose gel electrophoresis is a cornerstone technique for separating and analyzing nucleic acids, including DNA and RNA. Its widespread use in laboratories stems from its simplicity, efficiency, and effectiveness in separating fragments ranging from a few hundred base pairs to tens of thousands of base pairs. The process is based on the principle that negatively charged nucleic acid molecules migrate through a gel matrix when an electric current is applied.
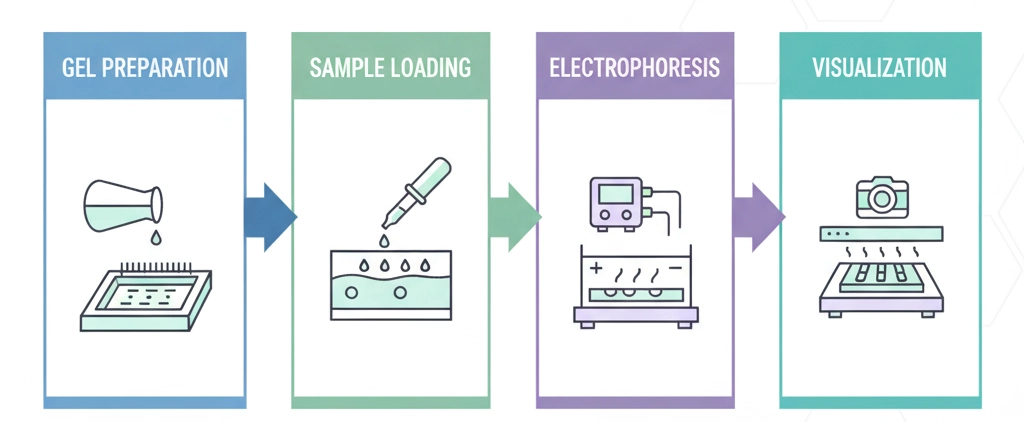
1. Gel Preparation and Casting: An agarose gel is prepared by dissolving agarose powder in a buffer solution, such as TAE (Tris-acetate-EDTA) or TBE (Tris-borate-EDTA). This mixture is heated until the agarose is fully dissolved and becomes a clear solution. The liquid gel is then poured into a casting tray equipped with a comb, which solidifies to form wells. The concentration of agarose is selected based on the size of the DNA fragments to be separated; higher concentrations provide better resolution for smaller fragments.
2. Sample Loading: Nucleic acid samples are mixed with a loading dye. The loading dye serves two primary functions: it provides a visual marker to track the sample's migration and contains a high-density agent (e.g., glycerol) that causes the sample to sink into the gel wells. A molecular weight ladder, or DNA marker, is loaded into one or more wells to serve as a reference for fragment size determination.
3. Electrophoresis Run: The gel tray is placed in an electrophoresis tank and submerged in the buffer. An electric current is applied, causing the negatively charged DNA fragments to migrate from the negative cathode toward the positive anode. During the run, smaller fragments move faster through the gel's pores, while larger fragments are retarded, resulting in a separation based on size. The technician monitors the migration of the loading dye to prevent the samples from running off the end of the gel.
4. Visualization and Analysis: Following the run, the gel is stained with a fluorescent dye that intercalates with nucleic acids, such as ethidium bromide or a safer alternative like SYBR Green. The gel is then placed on a UV transilluminator, and the separated DNA bands fluoresce, becoming visible. The size of the DNA fragments is determined by comparing their migration distance to that of the known fragments in the molecular weight ladder. This fundamental agarose gel electrophoresis is critical for a variety of applications, including PCR product verification, restriction enzyme digest analysis, and genotyping. This precise form of DNA analysis ensures the quality and quantity of nucleic acid samples prior to downstream applications like sequencing.
Optimizing Protein Separation with SDS-PAGE
While agarose gel electrophoresis is ideal for nucleic acids, the separation of proteins requires a different approach due to their variable charge and complex three-dimensional structures. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis, or SDS-PAGE, is the gold standard for separating proteins based on their molecular weight.
The SDS-PAGE technique overcomes the variable charge issue by treating protein samples with a denaturing agent, sodium dodecyl sulfate (SDS). This anionic detergent binds to the hydrophobic regions of proteins, causing them to unfold into a linear polypeptide chain. More importantly, SDS imparts a uniform negative charge-to-mass ratio on all proteins, effectively eliminating the influence of a protein's intrinsic charge on its migration rate. Consequently, protein separation in SDS-PAGE is based almost exclusively on size.
The SDS-PAGE Process:
Sample Preparation: Protein samples are mixed with a sample buffer containing SDS and a reducing agent (e.g., dithiothreitol or β-mercaptoethanol). The reducing agent breaks disulfide bonds, ensuring the complete denaturation of the protein subunits. The mixture is then heated, facilitating the full binding of SDS and linearization of the proteins.
Gel Casting: A polyacrylamide gel is used as the separation matrix. Unlike agarose, polyacrylamide is a chemical polymer with a highly uniform pore size, which provides superior resolution for proteins. The gel is typically cast in a vertical orientation and consists of two parts: a lower resolving gel and an upper stacking gel. The stacking gel concentrates the proteins into a narrow band before they enter the resolving gel, ensuring sharp, well-defined bands.
Electrophoresis Run: The prepared gel is placed in a vertical electrophoresis tank, filled with a running buffer. The treated protein samples and a pre-stained protein ladder are loaded into the wells. An electric current is applied, causing the negatively charged SDS-coated proteins to migrate from the cathode towards the anode. The smaller proteins travel faster through the gel matrix, while the larger ones are retarded, resulting in separation.
Visualization: After the run, the gel is typically stained with a dye such as Coomassie Blue, which binds to proteins, making the bands visible. The molecular weight of an unknown protein is then estimated by comparing its migration distance to the known molecular weights of the proteins in the pre-stained ladder. SDS-PAGE is a vital tool for numerous laboratory applications, including Western blotting, protein purity analysis, and protein expression studies. The precision of this protein analysis technique is fundamental for validating protein identity and function.
Exploring Advanced Electrophoresis Techniques and Essential Equipment
While standard gel electrophoresis methods are widely used, a variety of advanced techniques and sophisticated electrophoresis equipment have been developed to meet the demands of modern molecular biology and proteomics. These innovations offer enhanced resolution, higher throughput, and quantitative capabilities.
1. Capillary Electrophoresis (CE): CE is a high-resolution, automated technique that performs separations in a narrow, fused-silica capillary. In this method, the separation occurs entirely within the capillary, eliminating the need for a gel slab and allowing for rapid, high-throughput analysis. Samples are loaded electrokinetically, and the separation is driven by both an electric field and electro-osmotic flow. The separated molecules are detected by an in-line detector, providing a chromatogram-like output. CE is particularly useful for separating small molecules, ions, and large macromolecules like DNA and proteins, and is a staple in DNA sequencing and forensic analysis.
2. Two-Dimensional (2D) Gel Electrophoresis: 2D gel electrophoresis is a powerful technique for separating a complex mixture of proteins with very high resolution. The method separates proteins based on two independent properties in successive steps:
First Dimension (Isoelectric Focusing): of a protein's intrinsic charge on its migration rate. Consequently, protein separation in SDS-PAGE is based almost exclusively on size.
The SDS-PAGE Process:
Sample Preparation: Protein samples are mixed with a sample buffer containing SDS and a reducing agent (e.g., dithiothreitol or β-mercaptoethanol). The reducing agent breaks disulfide bonds, ensuring the complete denaturation of the protein subunits. The mixture is then heated, facilitating the full binding of SDS and linearization of the proteins.
Gel Casting: A polyacrylamide gel is used as the separation matrix. Unlike agarose, polyacrylamide is a chemical polymer with a highly uniform pore size, which provides superior resolution for proteins. The gel is typically cast in a vertical orientation and consists of two parts: a lower resolving gel and an upper stacking gel. The stacking gel concentrates the proteins into a narrow band before they enter the resolving gel, ensuring sharp, well-defined bands.
Electrophoresis Run: The prepared gel is placed in a vertical electrophoresis tank, filled with a running buffer. The treated protein samples and a pre-stained protein ladder are loaded into the wells. An electric current is applied, causing the negatively charged SDS-coated proteins to migrate from the cathode towards the anode. The smaller proteins travel faster through the gel matrix, while the larger ones are retarded, resulting in separation.
Visualization: After the run, the gel is typically stained with a dye such as Coomassie Blue, which binds to proteins, making the bands visible. The molecular weight of an unknown protein is then estimated by comparing its migration distance to the known molecular weights of the proteins in the pre-stained ladder. SDS-PAGE is a vital tool for numerous laboratory applications, including Western blotting, protein purity analysis, and protein expression studies. The precision of this protein analysis technique is fundamental for validating protein identity and function.
Exploring Advanced Electrophoresis Techniques and Essential Equipment
While standard gel electrophoresis methods are widely used, a variety of advanced techniques and sophisticated electrophoresis equipment have been developed to meet the demands of modern molecular biology and proteomics. These innovations offer enhanced resolution, higher throughput, and quantitative capabilities.
1. Capillary Electrophoresis (CE): CE is a high-resolution, automated technique that performs separations in a narrow, fused-silica capillary. In this method, the separation occurs entirely within the capillary, eliminating the need for a gel slab and allowing for rapid, high-throughput analysis. Samples are loaded electrokinetically, and the separation is driven by both an electric field and electro-osmotic flow. The separated molecules are detected by an in-line detector, providing a chromatogram-like output. CE is particularly useful for separating small molecules, ions, and large macromolecules like DNA and proteins, and is a staple in DNA sequencing and forensic analysis.
2. Two-Dimensional (2D) Gel Electrophoresis: 2D gel electrophoresis is a powerful technique for separating a complex mixture of proteins with very high resolution. The method separates proteins based on two independent properties in successive steps:
First Dimension (Isoelectric Focusing): Proteins are separated based on their isoelectric point (pI), the pH at which a protein has no net charge. This is performed using a pH gradient in a strip gel.
Second Dimension (SDS-PAGE): The strip gel from the first dimension is then placed on a standard SDS-PAGE gel and separated based on molecular weight, perpendicular to the first separation.
The result is a gel with proteins distributed as spots, providing a detailed map of a protein sample. 2D gel electrophoresis is a cornerstone of proteomics for comparing protein expression levels between different samples.
3. Essential Electrophoresis Equipment: The successful execution of any electrophoresis technique relies on a suite of specialized equipment. These include:
Power Supplies: These provide the constant voltage or current necessary to drive the migration of molecules. They are adjustable to accommodate different gel types and experimental requirements.
Gel Electrophoresis Systems: This includes the gel rigs (tanks and trays), combs, and casting platforms. Vertical systems are used for SDS-PAGE, while horizontal systems are standard for agarose gels.
Gel Imaging Systems: Modern systems use digital cameras and UV or chemiluminescent detection to capture high-quality images of the separated bands. They often include software for quantitative analysis of band intensity and molecular weight determination.
Capillary Electrophoresis Instruments: These are integrated, automated systems that combine the capillary, power supply, and detection components.
The integration of these advanced electrophoresis techniques and sophisticated electrophoresis equipment has significantly enhanced the precision, throughput, and analytical power of laboratory workflows.
Advancing Your Lab's Capabilities with Electrophoresis Mastery
Mastery of electrophoresis is a cornerstone of professional competence for laboratory personnel. This powerful technique provides a reliable and precise means for separating, identifying, and quantifying nucleic acids and proteins, which is foundational to a vast range of scientific and clinical applications. From the foundational principles of molecular migration in an electric field to the nuanced execution of techniques such as agarose gel electrophoresis and SDS-PAGE, a thorough understanding is non-negotiable for producing reliable scientific data.
The continuous evolution of electrophoresis equipment and the development of advanced methods like capillary and 2D gel electrophoresis underscore the dynamic nature of this field. By staying current with these innovations, laboratory professionals can not only improve the efficiency and accuracy of their work but also contribute more meaningfully to a wide spectrum of research, diagnostic, and industrial endeavors. The ability to troubleshoot, optimize, and execute these complex procedures with precision represents a critical professional skill set in the life sciences. Find the latest selection of electrophoresis equipment on LabX here.
FAQ
What is the fundamental difference between agarose gel electrophoresis and SDS-PAGE?
The primary difference lies in the molecules they are designed to separate and the principles of separation. Agarose gel electrophoresis is used for DNA analysis and RNA analysis, separating nucleic acids primarily based on their size. Because DNA and RNA molecules inherently possess a uniform negative charge-to-mass ratio, they migrate through the gel without the need for additional treatment. Conversely, SDS-PAGE is a specialized form of electrophoresis developed for separating proteins. Proteins have complex structures and variable charges, so the SDS detergent is used to denature them and impart a uniform negative charge, ensuring separation is based solely on their molecular weight.
Why is buffer selection so important in gel electrophoresis?
Buffer selection is crucial in gel electrophoresis as the buffer system performs multiple roles essential for successful separation. The buffer carries the electric current, acting as a conductor and maintaining a stable pH throughout the run. This stable pH ensures that the target molecules maintain their charge state, which is critical for consistent migration. For nucleic acids, buffers like TAE and TBE are commonly used to ensure the DNA remains negatively charged. For protein separation, the buffer also affects protein folding and the action of SDS, making the correct buffer choice fundamental for reproducibility and reliable results.
How does electrophoresis enable the accurate molecular weight determination of a sample?
Accurate molecular weight determination is achieved by running the sample alongside a molecular weight ladder, also known as a marker. This ladder consists of a mixture of molecules (either DNA or protein) of known, predetermined sizes. During the electrophoresis run, the molecules in the ladder separate according to their size, creating a series of distinct bands. The migration distance of the unknown sample band is then measured and compared to the migration distances of the known bands in the ladder. This comparison allows for a precise estimation of the unknown molecule's size, a vital step in characterizing samples.
What are the key considerations when selecting electrophoresis equipment for a laboratory?
The selection of electrophoresis equipment should be guided by the specific needs of the laboratory's applications. Key considerations include the types of molecules to be separated (DNA, RNA, proteins), the required resolution, and the desired throughput. For routine DNA and RNA work, a basic horizontal gel rig and power supply may suffice. Laboratories performing advanced protein studies or genomics will require more sophisticated equipment, such as vertical SDS-PAGE systems, high-resolution gel imagers, or automated capillary electrophoresis instruments. Choosing the right equipment ensures optimal performance, consistent results, and efficiency in laboratory workflows.
Jan 09, 2026
Product News
BRANDTECH Scientific Introduces the Transferpette® pro Micropipette: A New Twist on Comfort and Control
The latest addition to its trusted line of precision liquid handling instruments, engineered for comfort, control, and repeatable accuracy.
Jan 06, 2026
Featured, Technical Insight
New Research Frontiers Unlocked by Next-Generation Flow Cytometry
Next-generation flow cytometry systems using acoustic focusing are opening entirely new areas of research that were previously impractical or inaccessible with conventional flow cytometers.

Jan 05, 2026
Buying Guides
The Bottom Line: How Smart Microcentrifuge Tube Choices Impact Lab Efficiency and Cost
Innovative microcentrifuge tube solutions are more than just lab consumables—they’re strategic tools that can reduce costs, improve outcomes, and support long-term sustainability goals.
Jan 05, 2026
Buying Guides
Future-Proofing Your Lab with Innovative Microcentrifuge Tube Solutions
As laboratories evolve to meet the growing demands of modern science, even the most basic tools—like microcentrifuge tubes—are being reimagined.

Jan 05, 2026
Buying Guides
Microcentrifuge Tube Innovations: Driving Sustainable Change
Integrating next-generation consumables into standard workflows can lead to a more sustainable and responsible research environment.
Jan 05, 2026
Buying Guides
Single Use Plastics: How to Manage these Major Contributors to Lab Waste
Reducing lab waste isn’t just a sustainability issue—it’s a financial and operational one, too.
Jan 05, 2026
Buying Guides
Common Microcentrifuge Tube Challenges in Sensitive Molecular Workflows
Key factors that can impact the performance of microcentrifuge tubes for PCR, qPCR, and NGS and other sensitive applications
Jan 05, 2026
Buying Guides
How to Research the Right Microcentrifuge Tubes for Your Application
Selecting the right microcentrifuge tubes starts with understanding the specific demands of your application.
Jan 05, 2026
Technical Insight
The Best Confocal Microscopes of 2026
Discover the best confocal microscopes of 2026 for advanced imaging. Compare top models from Zeiss, Evident, and Nikon for speed, resolution, and budget.
Dec 24, 2025
Featured, Popular Products, Technical Insight
Emerging Mass Spectrometry Technologies to Improve Intraoperative Diagnostics
Advances in mass spectrometry may soon reshape surgery, improving diagnostic accuracy and impacting post-operative outcomes